基于活性的蛋白質組學分析(activity-based protein profiling,ABPP)是由Scripps研究所Cravatt教授團隊開發(fā),旨在利用不同反應活性的化學探針研究蛋白質的活性與功能。經過不斷的迭代發(fā)展,該技術已經被廣泛應用于共價小分子藥物靶點發(fā)現(xiàn)和先導化合物篩選領域。
以靶向半胱氨酸殘基的共價小分子為例,其技術路線即是利用靶向半胱氨酸的特異性化學探針為通用型探針,當共價小分子優(yōu)先占據靶點蛋白質的活性半胱氨酸殘基時,則會與通用型探針產生競爭,減弱探針的標記強度,結合定量蛋白質組學技術,能夠高效準確地鑒定共價小分子修飾蛋白質以及位點。2016年Cravatt教授團隊利用上述策略篩選共價小分子先導化合物,靶向“無藥可及”的蛋白質靶點[5]。該團隊首先構建了一個靶向半胱氨酸殘基的共價分子片段庫,它們具備與很多共價靶向藥物相同的丙烯酰胺或氯乙酰胺活性反應基團,結合化學蛋白質組學技術,作者全面探究了人類蛋白質組中半胱氨酸殘基與小分子片段的結合能力,在637個不同蛋白質上鑒定出758個能夠與小分子片段共價連接的活性半胱氨酸殘基,其中大部分都沒有在DrugBank數據庫中發(fā)現(xiàn),極大地完善了人類蛋白質組與結構各異的共價小分子相互作用數據網絡。該方法的運用加速了共價藥物先導化合物的篩選,在復雜蛋白質組水平上大規(guī)模發(fā)掘潛在配體結合口袋,為靶向“不可成藥”蛋白質提供了機會。該競爭性化學蛋白質組學技術還被應用于內源代謝產物小分子如hydroxynonenal(HNE)[6]、天然產物如具有抑癌活性的萜類化合物nimbolide的靶點發(fā)現(xiàn)中。
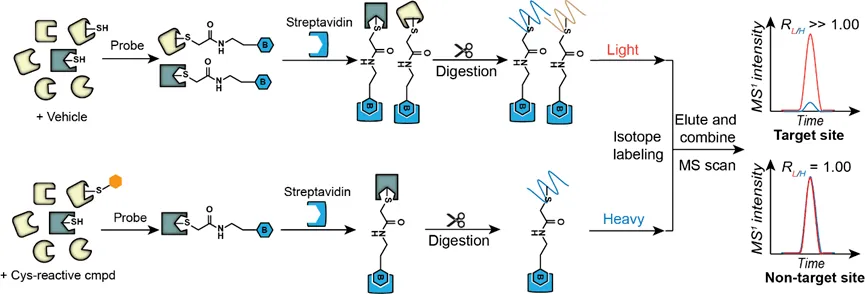
競爭性定量化學蛋白質組學技術鑒定共價藥物分子靶點和修飾位點技術路線示意圖近年來,靶向蛋白降解(targeted protein degradation, TPD),即利用藥物分子干預蛋白質的表達從而實現(xiàn)疾病治療的策略層出不窮,最受關注的要屬PROTAC(proteolysis targeting chimeras)藥物。PROTAC概念最早由Crews等人在2001年提出,該策略的核心思想是利用細胞內自有的泛素-蛋白酶體系統(tǒng)降低靶標蛋白的豐度,從而達到疾病治療的目的。同時,PROTAC藥物因其活性高、能夠靶向“不可成藥”靶點、克服藥物耐藥性等優(yōu)勢成為各大藥企的重點研發(fā)產品,輝瑞、拜耳、默沙東等國際制藥巨頭都在該方向上有所布局。類似的,同樣利用泛素-蛋白酶體系統(tǒng)的分子膠降解技術(molecular glue degraders)、利用內吞-溶酶體途徑的LYTAC技術(lysosome targeting chimeras)、利用自噬-溶酶體途徑的AUTAC(autophagy targeting chimera)和ATTEC(autophagosome tethering compounds)技術等。然而,這類分子研發(fā)過程中關鍵評價之一是能否特異性降解靶標蛋白,而不會導致細胞內其它蛋白質的豐度改變。定量蛋白質組學技術,即在蛋白質組水平,利用高分辨質譜定量比較不同樣品中相同蛋白質的含量,在該類藥物分子研發(fā)過程中扮演著不可或缺的角色,目前比較常用的定量蛋白組學策略是在肽段樣品制備過程中引入穩(wěn)定同位素標簽。質譜檢測時,來自不同樣品,同一氨基酸序列的肽段物理性質類似,具有相同的保留時間,電荷數,離子化效率和碎裂模式,但由于同位素標簽質量不同,從而表現(xiàn)出不同的荷質比,共流出的同一序列肽段離子強度之比即能夠定量反映不同樣品中同一蛋白質的含量。該策略不僅提高了了樣品制備、質譜數據采集等過程的效率,同時減少了樣品與樣品之間的實驗誤差,使結果更加穩(wěn)定、可信。
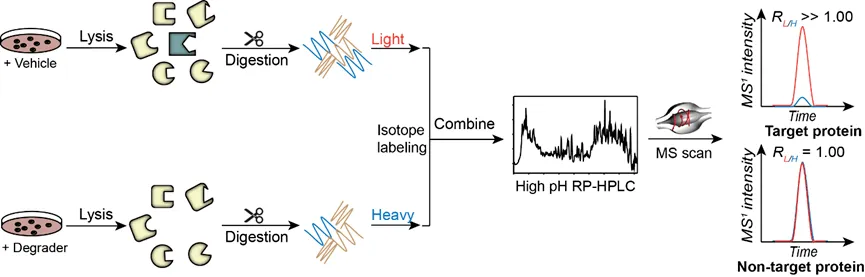
定量化學蛋白質組學技術鑒定PROTAC分子降解靶點技術路線示意圖隨著各學科的發(fā)展和交融,尤其是近年來化學生物學交叉學科的出現(xiàn),將藥物開發(fā)帶入了高速發(fā)展的新紀元,有望極大縮短新藥發(fā)現(xiàn)的周期。化學蛋白質組學技術的不斷發(fā)展,促進了細胞內代謝物、天然產物、合成藥物等小分子的靶點研究和全新藥物先導化合物的發(fā)現(xiàn),為小分子新藥研發(fā)注入了新的活力,使得原來被認為“不可成藥”的蛋白質有了成為新的治療靶點的可能。
